- #1
ponjavic
- 225
- 0
Hi
I am trying to run a FRAP (fluorescence recovery after photo-bleaching) experiment but I am struggling a little bit. For those not exposed to the technique it involves, for example, bleaching a spot in a specimen that is fluorescing rendering that spot dark in contrast.
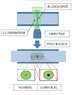
I have two separate beams going through my microscope which I suppose can be considered as a lens.
One beam is collimated and almost fills the lens. The other beam is diverging as to illuminate the surroundings to contrast with the dark central spot.
The reason why I'm even posing a question is that using a very thin specimen (<1 micron) I have no problems bleaching and identifying the spot. Once I have something larger (<100 microns) I am unable to do it.
This is my theory but I would like to back it up:
The bleach spot is obviously smaller than the depth of my channel. The illumination beam causes the dye to fluoresce above and below the bleach spot not allowing me to observe it. The solution to this would be using a confocal setup.
I've calculated the depth of focus to be approximately 10microns but I don't know if this is the depth of focus of what I can see (probably is) or if it's the depth of focus of what I am bleaching or if these are the same (should be I suppose but then I shouldn't be having problems).
Any input or if you want me to clarify something is welcome.
Aleks
Edit:
I also don't understand why a collimated laser beam when focused on a surface only covers about 5% of the field of view (as shown in the diagram the bleach spot is much smaller than the field of view).
Edit2:
Ok I just realized that focal point != field of view. The field of view will depend on where the binoculars are in relation to the objective. As my field of view is much larger than my focal point, so should my depth of view be much larger than the depth of focus (excuse the terminology) and thus the bleach spot will not be seen.
I am trying to run a FRAP (fluorescence recovery after photo-bleaching) experiment but I am struggling a little bit. For those not exposed to the technique it involves, for example, bleaching a spot in a specimen that is fluorescing rendering that spot dark in contrast.
I have two separate beams going through my microscope which I suppose can be considered as a lens.
One beam is collimated and almost fills the lens. The other beam is diverging as to illuminate the surroundings to contrast with the dark central spot.
The reason why I'm even posing a question is that using a very thin specimen (<1 micron) I have no problems bleaching and identifying the spot. Once I have something larger (<100 microns) I am unable to do it.
This is my theory but I would like to back it up:
The bleach spot is obviously smaller than the depth of my channel. The illumination beam causes the dye to fluoresce above and below the bleach spot not allowing me to observe it. The solution to this would be using a confocal setup.
I've calculated the depth of focus to be approximately 10microns but I don't know if this is the depth of focus of what I can see (probably is) or if it's the depth of focus of what I am bleaching or if these are the same (should be I suppose but then I shouldn't be having problems).
Any input or if you want me to clarify something is welcome.
Aleks
Edit:
I also don't understand why a collimated laser beam when focused on a surface only covers about 5% of the field of view (as shown in the diagram the bleach spot is much smaller than the field of view).
Edit2:
Ok I just realized that focal point != field of view. The field of view will depend on where the binoculars are in relation to the objective. As my field of view is much larger than my focal point, so should my depth of view be much larger than the depth of focus (excuse the terminology) and thus the bleach spot will not be seen.
Attachments
Last edited: