- #1
sludger13
- 83
- 0
I have some questions how various substituents are transfering benzene conjugated (π) electrons (i.e. activating/deactivating). As usual, no chemist provides an explanation.
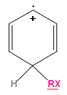
1)As I see, the first important thing when forming a carbocation during electrophilic aromatic substitution is to find out the part of the conjugated positive charge on the carbon that is binding the substituent. This seems clear for me, as this answers the resonance isomers.
2)As I see, the transfer of electron density just depends on the carbon positive charge. But there I'm not sure anymore. In benzene the carbon hybridization is to be (sp2), besides a substituent it could be maybe slightly crumpled. Thus, when (any part of) positive charge on the carbon, its hybridization shouldn't change, right?
It seemed to me probable that the electron density transfer (during conjugation, hyperconjugation) occurs because of different degree of orbital overlap because of different charge on an atom...
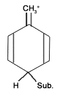
3)The second factor is the inductive effect. Wikipedia says a donation of electron density into conjugated system is via inductive effect. Does this mean a hyperconjugation? I have no idea how inductive effect could affect the electron density without hyperconjugation (e.g. with deactivating trifluorinemethyl R-CF3 the transfer from the benzene is greater with greater carbon positive charge).
4)Also, when (sp2) substituent (for example R-NH2), the hyperconjugation shouldn't occur. Is it true?
1)As I see, the first important thing when forming a carbocation during electrophilic aromatic substitution is to find out the part of the conjugated positive charge on the carbon that is binding the substituent. This seems clear for me, as this answers the resonance isomers.
2)As I see, the transfer of electron density just depends on the carbon positive charge. But there I'm not sure anymore. In benzene the carbon hybridization is to be (sp2), besides a substituent it could be maybe slightly crumpled. Thus, when (any part of) positive charge on the carbon, its hybridization shouldn't change, right?
It seemed to me probable that the electron density transfer (during conjugation, hyperconjugation) occurs because of different degree of orbital overlap because of different charge on an atom...
3)The second factor is the inductive effect. Wikipedia says a donation of electron density into conjugated system is via inductive effect. Does this mean a hyperconjugation? I have no idea how inductive effect could affect the electron density without hyperconjugation (e.g. with deactivating trifluorinemethyl R-CF3 the transfer from the benzene is greater with greater carbon positive charge).
4)Also, when (sp2) substituent (for example R-NH2), the hyperconjugation shouldn't occur. Is it true?