FoxOne said:
1. It can't be right. The hybridorbital (sp3) is a linear combination of the wavefunctions of the atomic orbitals.
Good... you are making the first step to understanding what we are talking about. And now for the interesting question: If hybrid orbitals are linear combinations of atomic orbitals, and molecular orbitals are linear combinations of hybrid orbitals... what is stopping you from expressing one and the same set of final molecular orbitals with DIFFERENT sets of intermediate hybrid orbitals? And what is stopping you from skipping the hybrid orbitals altogether and expressing the molecular orbitals directly as linear combinations of atomic orbitals? Or as linear combination of functions which are not even atomic orbitals themselves, but simply allow expressing the AOs/MOs efficiently?
[...]And I think you have an imagination problem. Because you try to tell me that the one-electron orbital wave functions of oxygen (s- and p-atomic orbitals) remains constant after bringing the hydrogen s-atomic orbitals close to it. This can't be.
No one said anything to that effect. What was said is that you can express bonding situation by invoking the atomic orbitals directly, rather than hybrid orbitals (or choosing various different sets of different hybrid orbitals: here sp2 and sp3, for example). And this is obviously true, as the MOs can be expressed over AOs rather than hybrids.
By the way, what you described is polarization, not hybridization. And, of course, strictly speaking, atomic orbitals in molecules are not real (i.e., physically observable) either...
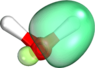
2. Your pictures directly show the hybridization mentioned in the literature in point 1. These are sp2 b1 hybrid bonds.
That is one way of seeing it. It shows that, just as DrDu said earlier, the two lone pairs need not be considered equivalent and that the O need not be described as sp3 hybridized. It thus contradicts your statements from post #5, where you incorrectly claimed that this cannot be true.
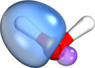
By the way: I attached another set of orbitals to the current post. The set of orbitals attached here and the set in #6 are physically equivalent[1] and describe the same many-body wave function. Any physically observable quantity computed from either set of orbitals will come out equal to within numerical precision (i.e., 10-14 decimals digits in double precision arithmetic). Where are your sp3 vs sp2 hybrids now? Muhahaha! (<- evil laugh). Mind blown? 8).
And before you tell me that I am wrong and this cannot be true: No, I am not wrong, and yes, this is very much true.
[1] Both of them represent the same DF-RKS/PBE/def2-TZVP wave function. The ones in #6 are Intrinsic Bond Orbitals (IBOs) made in IboView (which can be downloaded at
www.iboview.org if you want to see it for yourself), the ones attached here are Boys orbitals. Since you mentioned Hartree-Fock: HF orbitals would be quantitatively somewhat different, but would look visually indistinguishable from the DFT orbitals, and they could be localized in the same way.
3. I have a basic understanding of quantum mechanics, you are not the only one who studied, and i know what a orbital is. But you try to explain molecular bondings with atomic orbitals and this is completely wrong. Can you tell me why there are numerous groups working on Hartree-Fock methods to calculate molecular ORBITALS?
Pro tip: This is a semi-anonymous internet forum. Some people take the time to answer 100s of highly technical questions about very particular topics in science and research. Ever wondered what kind of people might do this? What kind of day job those people might have?...
If I
guarantee you that I know all the gory details about electronic structure theory, and I know
exactly how it is implemented in computer programs---what do you think? How could I be in a position where I can
guarantee (hint, hint) that I know
exactly (hint, hint) how it works?
4. The only new method which is working without orbitals is the so called multi-configurational self-consistent field method. This method is working with the linear combination of state functions because sometimes Hartree-Fock or density functional theory methods bring wrong or unrealistic results.
1. MCSCF is not a new method.
2. MCSCF does use orbitals. Orbitals are what the CSFs or determinants are made of.
3. There are *many* electronic structure methods which are not based on orbitals (e.g., various kinds of real-space quantum Monte-Carlo methods, various first-quantized variational wave functions, "explicitly correlated" wave functions) or which invoke orbitals, but only for computational reasons, not to express wave functions (e.g., various geminal methods like the anti-symmetrized geminal power).
4. None of those have *anything* to do with the topic we had been discussing. The qualitative electronic structure of most molecules, especially of H2O, is well described by a single determinant, as used in Hartree-Fock and Kohn-Sham methods. These are perfectly adequate methods to base discussions of hybridization on.
5. Your argument with the computer programs is very funny. Orbital means one-electron orbital wave function. So when a program use wave functions it uses orbitals and when the program made a linear combination of this atomic orbitals we will get another orbital. Hope i can show you that you the confused one.
8).
No, but seriously: You need to work on your attitude. Both in science and real life it is *very* important that one has not only a good understanding of how things work, but also to which degree what one thinks is "knowledge" can be trusted. You just happened to say a number of very, very wrong things in this thread, coupled with implying that your conversation partners are the ones who do not know what they are talking about (pro tip: they do know). Now, as mentioned, this is a semi-anonymous internet forum so this particular thread will most likely not come back to haunt you in the future. But I would *strongly* advise not to pull off anything like this in your day job or other real-life situations. It can backfire. Hard.
And with your point that hybridization is nothing we can experimental proof i disagree.
One thing I like about physical facts is that they do not go away just because someone does not agree with them.