danielw96
- 2
- 0
- TL;DR
- I created a Lindbladian for a system of coupled orbitals, and now I want to calculate the interacting non equilibrium Greens function, using the Lehmann representation.
Hey :)
Firstly I want to thank everyone who takes their time to read through this post and who tries to help me.
So the issue is the following:
I wrote a python code that creates a Lindbladian, and I wanted to try to calculate the Greens function using the Lehmann representation.
For the easiest case of one site with no spin this works out. I know that the result is correct, because I used eq. 36 from the following paper: https://arxiv.org/pdf/1312.4586.pdf (this is the paper I will refer to with eq. numbers), which is for the case of non interacting particles, and compared it with the one i get from eq. 43 (Lehmann representation, where I only used the first some since I just wanted to calculate G>+ here) which also includes the case of interaction. Eventually I want to use this for the case of two spins per site, so that's why I am using the formula that also captchers on site interaction between two electrons, but for simplicity reasons I just went to the next step after a one site and no spin case i.e. two sites with no spin that are not coupled.
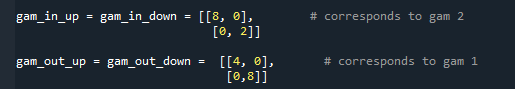
So I basically set every parameter to zero except for gamma_1 and gamma_2, (eq. 10b) which i individually gave values for each of the two sites. (screenshot). In this case one would expect the result I would get for a single site no spin level with given gamma_1 and gamma_2 values for each of the two sites.
Well and now we get to the reason for my post -> This doesn't work. And even worse if I change the gamma_1 or gamma_2 value for site number one then the result of the greens function calculated from the Lehmann representation also changes for site number two (and vice versa) which makes absolutely no sense, since the two sites are NOT coupled!
However I cannot find the issue with my code, I spent 40 hours in looking for the error, but the code does as far as I could test it everything it's supposed to do.
The Lindbladian itself should be correct, since I not only find the steady state, but also I get consistent results for <N>, as well as the current into and out of each site. I checked the result of <N> for decoupled sites were each site has some gamma_1 and gamma_2 values and it gave me individually what one would expect for single sites i.e. gamma_2/(gamma_1+gamma_2).
I calculated several matrix elements of the Lindbladian in the N - N^tilde = 1 basis (which i need for calculating the greens function) by hand, and all of them were correct.
Also I checked the calculation of the individual terms of the sum of the Greens function in Lehmann representation as shown in eq. 43, and also this function does exactly what it should. -> So what I did was that I let the first annihilation operator act on the right eigenstate of L_+1. This right eigenstate is a superposition of the N - N^tilde = 1 basis and so I checked which states of this basis survives the annihilation. Then my program checks where the corresponding states in the N - N^tilde = 0 basis are, so that I know which components of each L_+1 right eigenstate I have to multiply with the so called left vacuum.
Pretty much the same I did with the creation operator i.e. I let it act on the left eigenstates where it annihilates a particle in each N - N^tilde = 1 state of which each left eigenstates is build up, and multiplied with the corresponding values of the steady state.
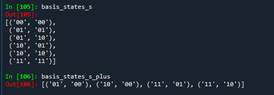
You can see in the screenshot called "bases" the two bases N - N^tilde = 0 and = 1, and then in screenshot named "corresponding states" you can see which states my code says are matching after annihilating a particle in the first site, and as you can see (starting numbering from 0) the 1st, 2nd and 3th state of the +1 basis survive and are then |00>|00>, |01>|01>, and |01>|10>, which correspond to 0th, 1st and 2nd state of the 0 basis.
You can find my code attached, the functions of relevance here are called "get_just_L_s", and "get_G_greater", and they are in the file called "main_functions_6_may_1_site" (name is misleading, but originally I planned to use it for a single site, however i then extended two two but can be arbitrarelly choosen)
I hope my explanation of how I proceeded was kinda clear, if not do not hesitate to ask!
Also I am not sure if I gave enough information for you to make a judgement on what might be wrong so if you require anything more just let me know and I'll do my best to provide it.
Firstly I want to thank everyone who takes their time to read through this post and who tries to help me.
So the issue is the following:
I wrote a python code that creates a Lindbladian, and I wanted to try to calculate the Greens function using the Lehmann representation.
For the easiest case of one site with no spin this works out. I know that the result is correct, because I used eq. 36 from the following paper: https://arxiv.org/pdf/1312.4586.pdf (this is the paper I will refer to with eq. numbers), which is for the case of non interacting particles, and compared it with the one i get from eq. 43 (Lehmann representation, where I only used the first some since I just wanted to calculate G>+ here) which also includes the case of interaction. Eventually I want to use this for the case of two spins per site, so that's why I am using the formula that also captchers on site interaction between two electrons, but for simplicity reasons I just went to the next step after a one site and no spin case i.e. two sites with no spin that are not coupled.
So I basically set every parameter to zero except for gamma_1 and gamma_2, (eq. 10b) which i individually gave values for each of the two sites. (screenshot). In this case one would expect the result I would get for a single site no spin level with given gamma_1 and gamma_2 values for each of the two sites.
Well and now we get to the reason for my post -> This doesn't work. And even worse if I change the gamma_1 or gamma_2 value for site number one then the result of the greens function calculated from the Lehmann representation also changes for site number two (and vice versa) which makes absolutely no sense, since the two sites are NOT coupled!
However I cannot find the issue with my code, I spent 40 hours in looking for the error, but the code does as far as I could test it everything it's supposed to do.
The Lindbladian itself should be correct, since I not only find the steady state, but also I get consistent results for <N>, as well as the current into and out of each site. I checked the result of <N> for decoupled sites were each site has some gamma_1 and gamma_2 values and it gave me individually what one would expect for single sites i.e. gamma_2/(gamma_1+gamma_2).
I calculated several matrix elements of the Lindbladian in the N - N^tilde = 1 basis (which i need for calculating the greens function) by hand, and all of them were correct.
Also I checked the calculation of the individual terms of the sum of the Greens function in Lehmann representation as shown in eq. 43, and also this function does exactly what it should. -> So what I did was that I let the first annihilation operator act on the right eigenstate of L_+1. This right eigenstate is a superposition of the N - N^tilde = 1 basis and so I checked which states of this basis survives the annihilation. Then my program checks where the corresponding states in the N - N^tilde = 0 basis are, so that I know which components of each L_+1 right eigenstate I have to multiply with the so called left vacuum.
Pretty much the same I did with the creation operator i.e. I let it act on the left eigenstates where it annihilates a particle in each N - N^tilde = 1 state of which each left eigenstates is build up, and multiplied with the corresponding values of the steady state.
You can see in the screenshot called "bases" the two bases N - N^tilde = 0 and = 1, and then in screenshot named "corresponding states" you can see which states my code says are matching after annihilating a particle in the first site, and as you can see (starting numbering from 0) the 1st, 2nd and 3th state of the +1 basis survive and are then |00>|00>, |01>|01>, and |01>|10>, which correspond to 0th, 1st and 2nd state of the 0 basis.
You can find my code attached, the functions of relevance here are called "get_just_L_s", and "get_G_greater", and they are in the file called "main_functions_6_may_1_site" (name is misleading, but originally I planned to use it for a single site, however i then extended two two but can be arbitrarelly choosen)
I hope my explanation of how I proceeded was kinda clear, if not do not hesitate to ask!
Also I am not sure if I gave enough information for you to make a judgement on what might be wrong so if you require anything more just let me know and I'll do my best to provide it.