chagular
- 5
- 0
For a real gas (non-ideal gas) in a reversible process, the way to calculate [itex]\Delta{S}[/itex] should also be independent to path simply because entropy is a state function.
However, I got strange solution while taking different path.
Here is the condition:
1. Equation of state (EOS) for the real gas could be arbitrary (but actually not ideal)
2. The constant volume heat capacity is only the function of temperature or a constant, i.e., [itex]C_V=f(T)[/itex] only.
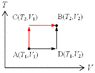
3. State changes from [itex]A(T_1,V_1)[/itex] to [itex]B(T_2,V_2)[/itex].
As illustrated (attached figure). Basically, there're two simple paths for calculation.
(1) The black route: A-D-B
(2) The red route: A-C-B
The total differential of entropy as function of [itex]T[/itex] and [itex]V[/itex] is:
[itex]dS=\frac{C_V}{T}dT+\left(\frac{\partial P}{\partial T}\right)_VdV[/itex].
Both entropy change in constant-V process A-C and D-B are identical:
[itex]\Delta{S}=\int^{T_2}_{T_1}\frac{C_V}{T}\,dT[/itex].
But I can't prove that the rest two constant T process paths have same contribution as encountering a real gas:
(1) for A-D (in black route)
[itex]\Delta{S_{AD}}=\int^{V_2}_{V_1} \left.\left(\frac{\partial P}{\partial T}\right)_V\right|_{T_1}\,dV[/itex]
(2) for C-B (in red route)
[itex]\Delta{S_{CB}}=\int^{V_2}_{V_1} \left.\left(\frac{\partial P}{\partial T}\right)_V\right|_{T_2}\,dV[/itex]
It's obvious that [itex]\Delta{S_{AD}}[/itex] is not identical to [itex]\Delta{S_{CB}}[/itex] if [itex]\left(\frac{\partial P}{\partial T}\right)_V[/itex] is function of both V and T. Such a situation could happen for almost all real gas.
The result obtained above is opposed to the fundamental knowledge: entropy is a state function. Regardless of the integration difficulty that might encounter as we could always use numerical integration especially when analytical EOS is available. What is wrong with my derivation?
However, I got strange solution while taking different path.
Here is the condition:
1. Equation of state (EOS) for the real gas could be arbitrary (but actually not ideal)
2. The constant volume heat capacity is only the function of temperature or a constant, i.e., [itex]C_V=f(T)[/itex] only.
3. State changes from [itex]A(T_1,V_1)[/itex] to [itex]B(T_2,V_2)[/itex].
As illustrated (attached figure). Basically, there're two simple paths for calculation.
(1) The black route: A-D-B
(2) The red route: A-C-B
The total differential of entropy as function of [itex]T[/itex] and [itex]V[/itex] is:
[itex]dS=\frac{C_V}{T}dT+\left(\frac{\partial P}{\partial T}\right)_VdV[/itex].
Both entropy change in constant-V process A-C and D-B are identical:
[itex]\Delta{S}=\int^{T_2}_{T_1}\frac{C_V}{T}\,dT[/itex].
But I can't prove that the rest two constant T process paths have same contribution as encountering a real gas:
(1) for A-D (in black route)
[itex]\Delta{S_{AD}}=\int^{V_2}_{V_1} \left.\left(\frac{\partial P}{\partial T}\right)_V\right|_{T_1}\,dV[/itex]
(2) for C-B (in red route)
[itex]\Delta{S_{CB}}=\int^{V_2}_{V_1} \left.\left(\frac{\partial P}{\partial T}\right)_V\right|_{T_2}\,dV[/itex]
It's obvious that [itex]\Delta{S_{AD}}[/itex] is not identical to [itex]\Delta{S_{CB}}[/itex] if [itex]\left(\frac{\partial P}{\partial T}\right)_V[/itex] is function of both V and T. Such a situation could happen for almost all real gas.
The result obtained above is opposed to the fundamental knowledge: entropy is a state function. Regardless of the integration difficulty that might encounter as we could always use numerical integration especially when analytical EOS is available. What is wrong with my derivation?